Notre équipe s’est développée depuis plusieurs années autour de l’étude de syndromes rares de prédisposition tumorale, en particulier les neurofibromatoses. Nous étudions plus particulièrement, d’une part les mécanismes génétiques expliquant ces syndromes et leurs présentations cliniques variables et d’autre part la génétique des tumeurs bénignes ou cancéreuses associées à ces syndromes, ainsi que leur ciblage thérapeutique.
Nous nous intéressons en particulier :
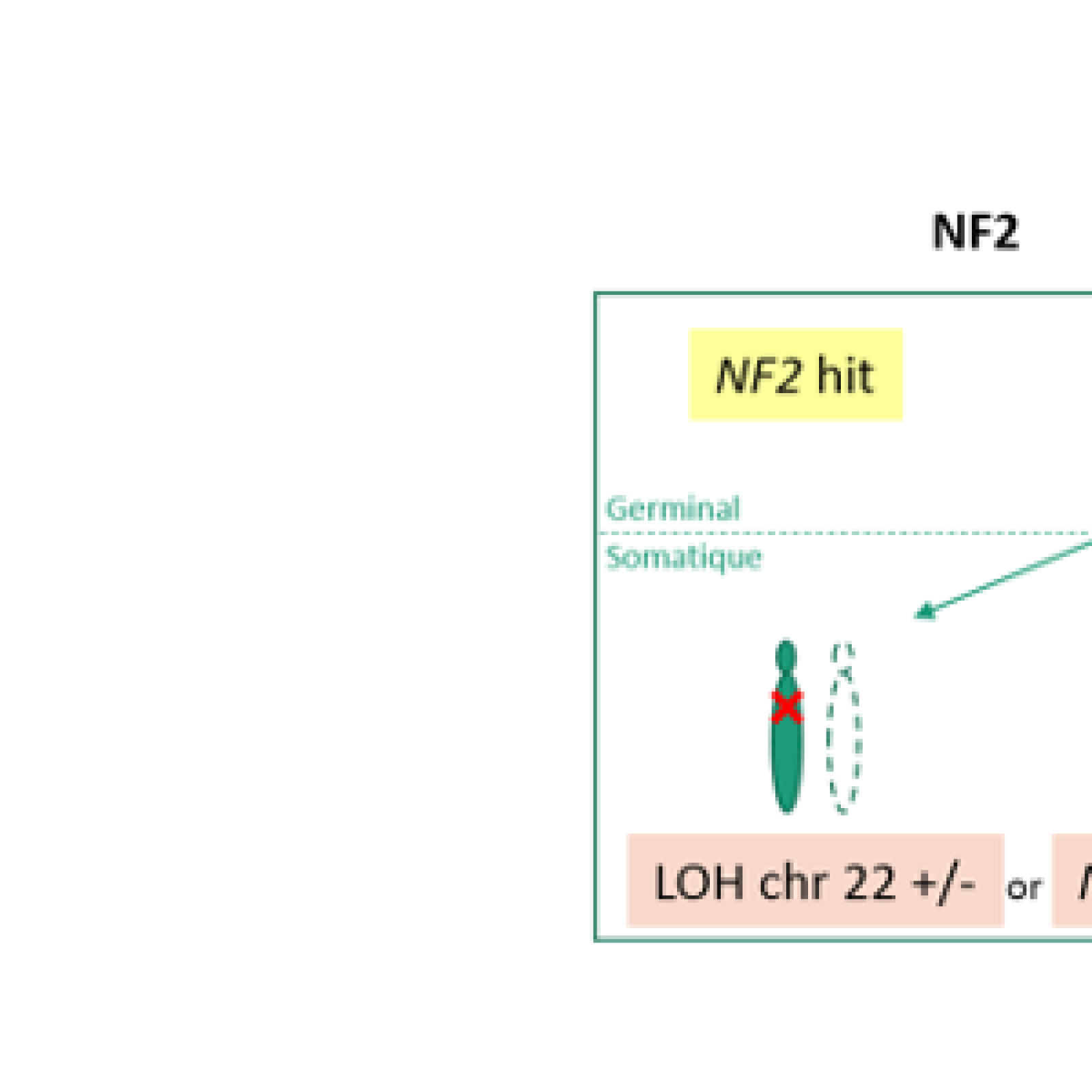
- A l’étude des mécanismes génétiques et épigénétiques de syndromes rares de prédisposition tumorale. Nous avons pour objectif de mieux comprendre les bases génétiques de l’expressivité variable (étude de corrélations génotype-phénotype, identification de gènes modificateurs et développement d’approches de génomique fonctionnelle à haut débit) et l’hétérogénéité génétique de syndromes rares de prédisposition tumorale, incluant la neurofibromatose de type 1 (NF1), la neurofibromatose de type 2 (NF2) et la schwannomatose. Plus récemment, l’équipe a initié une activité de recherche dédiée à des syndromes de prédisposition aux tumeurs digestives, incluant le syndrome de Peutz-Jeghers et la Polypose adénomateuse associée aux ADN polymérases (PPAP) et développe des tests génomiques fonctionnels à haut-débit pour interpréter les mutations faux-sens du gène POLE impliqué dans la PPAP ;
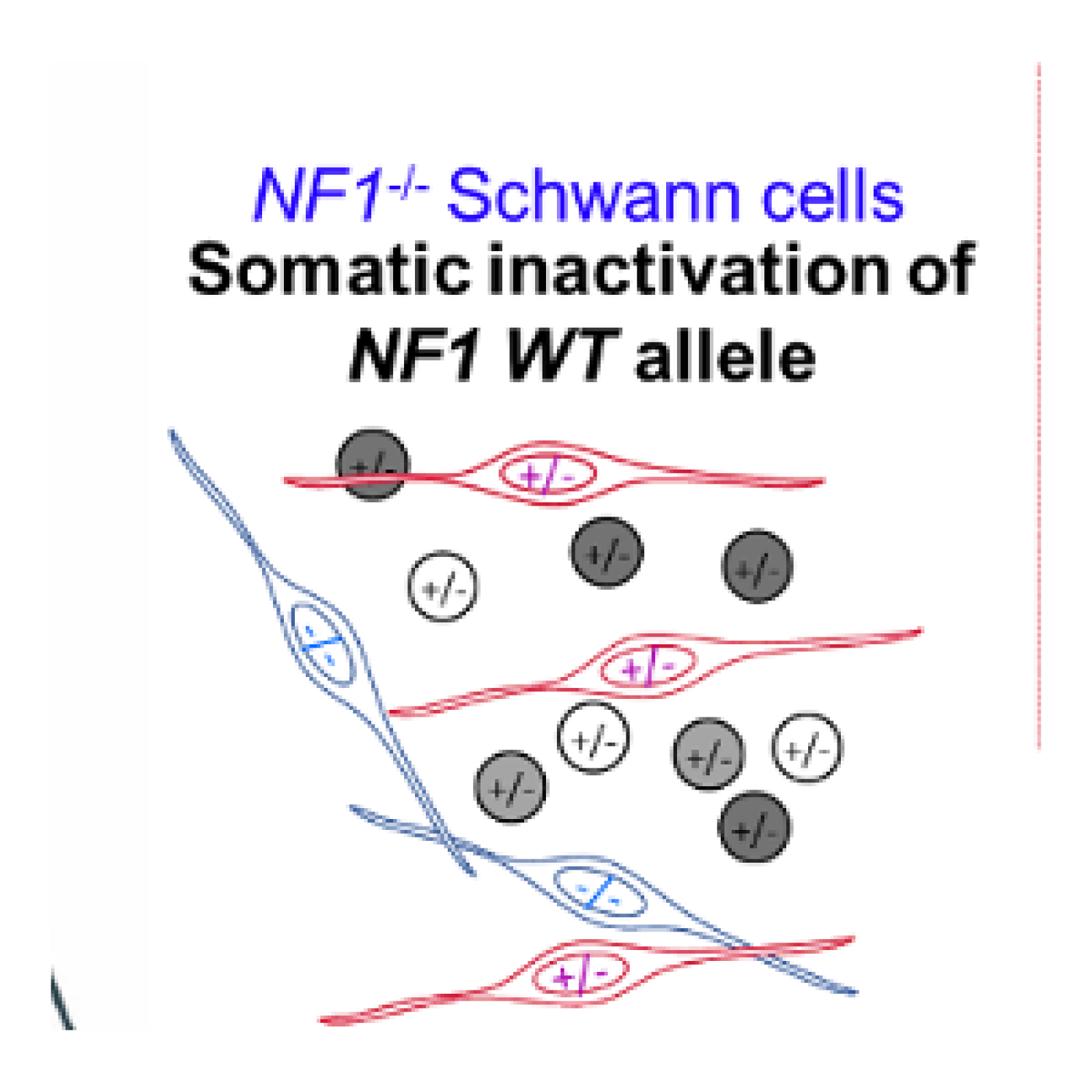
- A l’étude fonctionnelle des tumeurs associées à la NF1/NF2 ou impliquant le gène NF1/NF2, pour la caractérisation des acteurs majeurs de leur développement et la recherche d’approches thérapeutiques, notamment par l’identification de cibles d’intérêt par des cribles pharmacologiques et génétiques.