L’équipe étudie les relations entre le métabolisme énergétique et des caractères plus complexes au niveau de la cellule ou de l’organisme entier.
La respiration cellulaire fournit l’énergie nécessaire à la survie, l’activité et la croissance des mammifères. Cette respiration produit du gaz carbonique (CO2) et de l’eau. Une simplification consiste à diviser ce processus en deux étapes :
- l’oxydation des substrats (glucides, lipides, protéines) en CO2 (étapes métaboliques),
- la conversion de cette énergie d’oxydation en une forme utilisable par la cellule, la molécule d’ATP, avec consommation d’oxygène et production d’eau (bioénergétique).
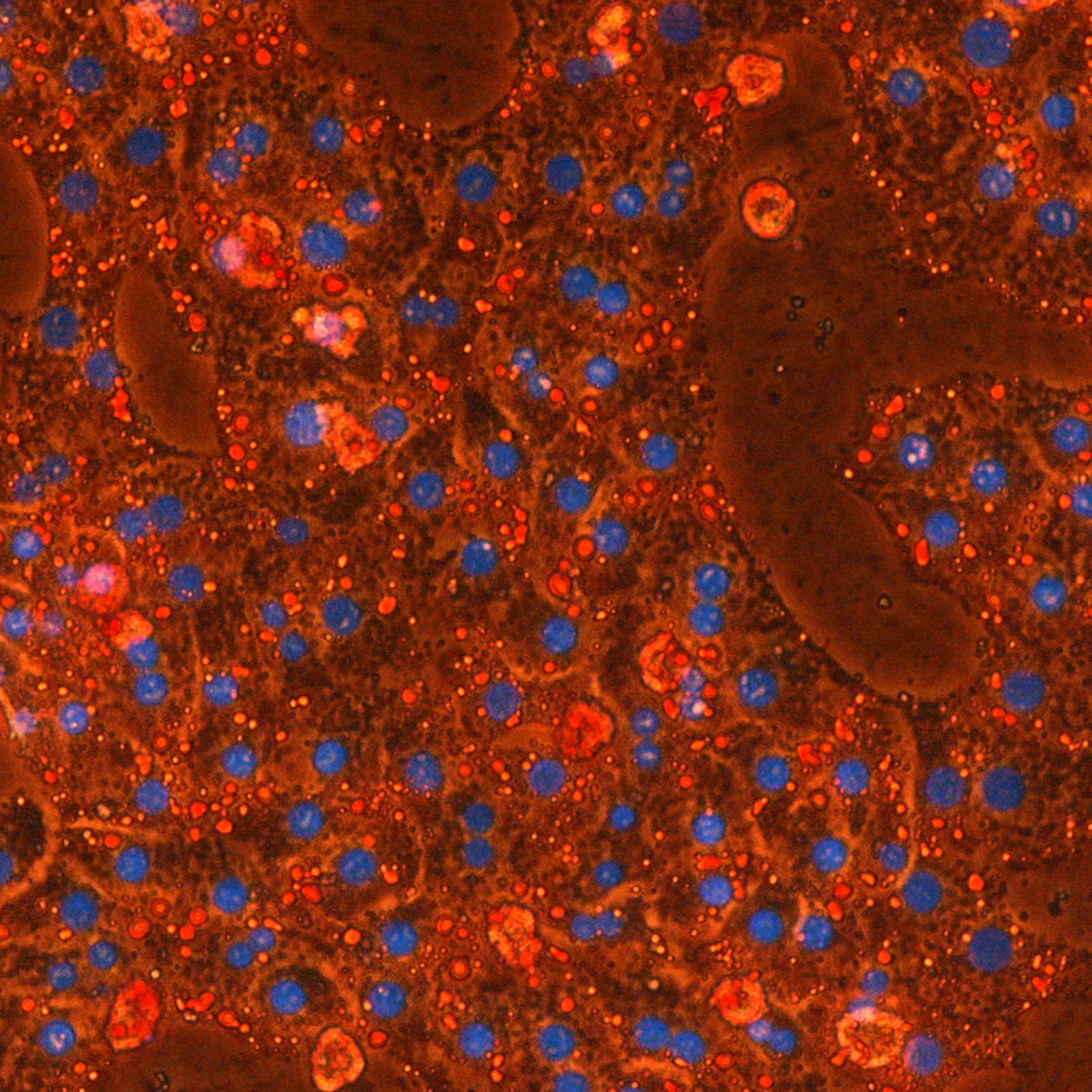
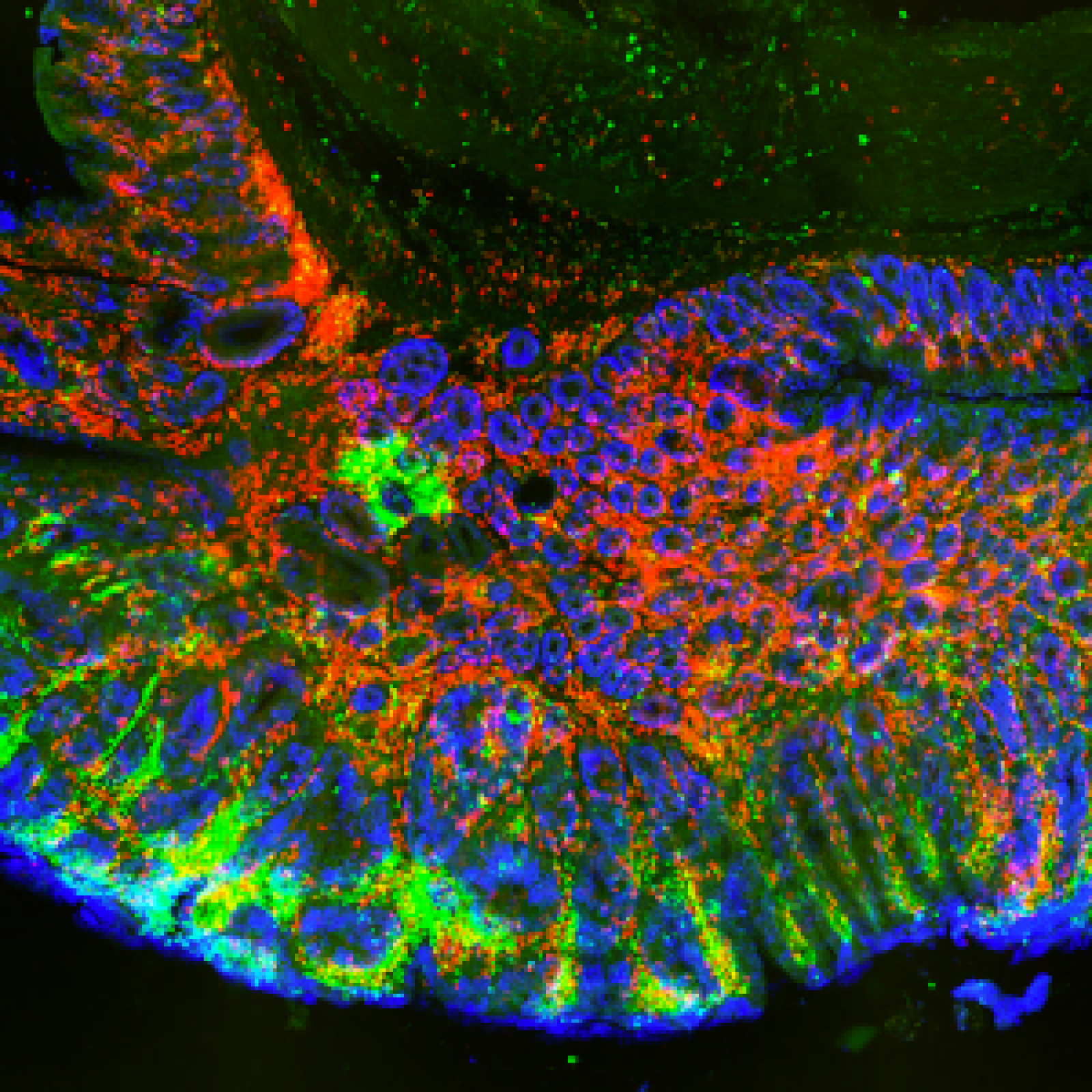
Les étapes métaboliques ont lieu en majorité à l’intérieur des mitochondries, et la production d’ATP est essentiellement due à l’activité de complexes enzymatiques insérés dans la membrane interne (phosphorylation oxydative). Une petite quantité d’ATP est produite par certaines étapes métaboliques. Localement et/ou transitoirement, elles peuvent devenir la seule source énergétique de la cellule. C’est le cas de la glycolyse dont deux étapes sont génératrices d’ATP et qui, en conditions anaérobies, est associée à la fermentation lactique.
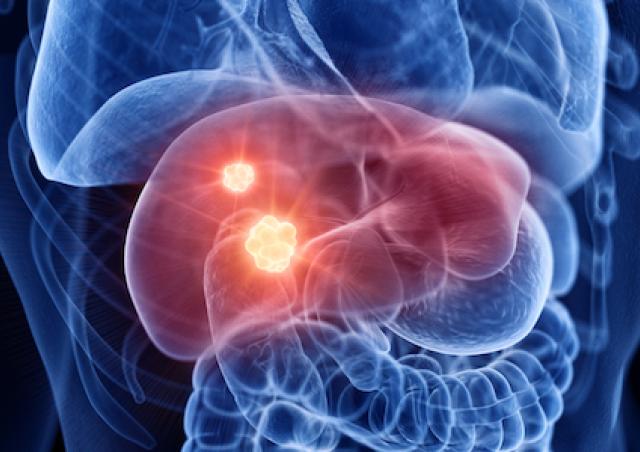
Des variations du métabolisme énergétique accompagnent diverses pathologies (obésité, diabète, cancer, maladies neurodégénératives) et sont aussi corrélées au vieillissement. La question est de déterminer si ces modifications sont
- des marqueurs,
- des facteurs aggravants,
- ou directement causatives d’une pathologie.
En ce qui concerne le cancer, le débat existe depuis la théorie de Warburg associant glycolyse (à la base de la détection des tumeurs par PET scan) et phénotype cancéreux. Les relations entre métabolisme énergétique et cancer sont devenues le sujet principal de l’équipe. L’équipe développe aussi des projets en étroite relation avec son expertise mitochondriale, souvent liés à des collaborations académiques ou avec des partenaires industriels.